 These kinds of multiplets,
of any complexity met with in practice, may be displayed
using the Windows program
Mltiplet
available for download from the present website
These kinds of multiplets,
of any complexity met with in practice, may be displayed
using the Windows program
Mltiplet
available for download from the present website
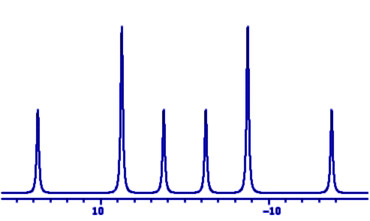
Students normally meet NMR spin-spin coupling constants J
as splittings in doublet, triplet etc. multiplets, with
relative intensities of their peaks determined by
Pascal's triangle, and symmetrically distributed about the
centre of the multiplet.
These kinds of multiplets,
of any complexity met with in practice, may be displayed
using the Windows program
Mltiplet
available for download from the present website
These multiplets are called 'first order' because the frequencies of individual peaks may be calculated from the chemical shift ν and the couplings J by first order arithmetic, not involving squares or higher powers of the parameters.
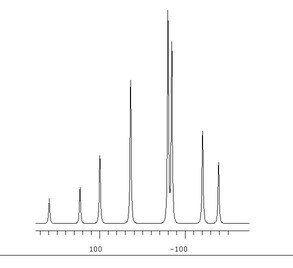
In reality, this is strictly true only for some heteronuclear couplings, e.g. between 1H and 13C. When homonuclear couplings, e.g. 1H—1H or 31P—31P, are present, some quadratic or higher order arithmetic may be involved in finding values of J and ν from the spectrum. In general these are called 'non–first–order' systems.
Non–first–order systems may approximate to first order ones if δAB, the difference in chemical shift in Hz between nuclei A and B, is large compared with JAB, the coupling between them, e.g. J/δ<0.05. In the absence of molecular symmetry, which may cause zero difference in chemical shift, and with increasingly high operating frequencies which make δAB bigger when expressed in Hz, the first order approximation is often used in organic chemistry. Observed homonuclear splittings are often reported as couplings. If available software for fitting the spectrum to parameters can cope with the size of the spin system, then values of ν and J obtained from a fit will always be preferable, if only because the fitting routine will average out experimental error in an overdetermined case.
Traditional programs such as Numarit, a convenient Windows implementation of which may be downloaded from the present website, require input of starting values of chemical shifts ν and couplings J, from which to start an iterative fit of the experimental spectrum. Where J/δ is small, interpreting splittings as couplings, and multiplet midpoints as chemical shifts, may provide an adequate starting point. Where J/δ is larger, or even infinity because of symmetry, this is not possible, and hand analysis must be done.
Here is a progressive set of exercises to be done by students using hand calculators, which aims to teach the art of this hand analysis for simple systems of up to four spin–½ nuclei. Only basic familiarity with solving pairs of linear simultaneous equations is required.
In contrast to traditional courses in this area, which use examples from organic 1H NMR, this one uses inorganic 31P NMR examples, mostly from the author's own research. In his experience, practising organic synthetic chemists analyse non-first-order spectra relatively rarely, whereas in identification of more symmetric inorganic molecules, and particularly in analysing satellite spectra caused by low-abundance isotopes, it is much more necessary.
The exercises were run originally as a taught class, in which skilled academic staff or postgraduates would help students with their difficulties. However, they are now best used with Numarit, into which you may input your found shifts and couplings, to find out whether they correctly exactly reproduce the provided spectra.
The Numarit package will provide also all of the final answers to the exercises, so if you have gone wrong it should not be too difficult to work back to the values of intermediate results such as N and L, to spot the source of the error. So as not to spoil the exercises for any teacher of NMR who wishes to use them for in-person teaching, these intermediate 'answers' will not be published here. The most common point of error is lack of care or poor reasoning in assigning a sign to a parameter measured as a splitting.
The course is available as a .pdf file for Acrobat Reader at
https://teaching.ncl.ac.uk/chemmodels/teaching/drylabs/nfonmr.pdf
or as a Word .docx file, at
https://teaching.ncl.ac.uk/chemmodels/teaching/drylabs/nfonmr.docx