ORBITAL for Web Browsers
Program ORBITAL shows coloured contour plots of functions which represent
simple atomic or molecular orbitals. It is intended to be used by students
learning about molecular orbital theory in chemistry courses at an elementary
undergraduate level.
It was written by Dr. Bruce W. Tattershall, Lecturer in Chemistry at
Newcastle University, England.
Introduction Contents

Preface
- ORBITAL for Web Browsers is a translation into HTML5.0 and
JavaScript of the stand-alone Windows application
ORBITAL for Windows
- ORBITAL for Windows works well in PCs running versions of
Windows including Windows 95, Windows XP, Windows 7, and
up to and including 64-bit Windows 10, and is still available for
free download
but its limitation is that it will run only in Windows-based
devices
- ORBITAL for Web Browsers attempts to avoid this limitation
by using your web browser's interface to your device's
operating system to provide Windows-like controls and display
facilities, as well as the required calculating ability
- There are many versions of web browsers in many operating systems,
but not all will support ORBITAL for Web Browsers
- If the ORBITAL for Web Browsers web page comes up looking
like the screen shot shown at the top left of this help page,
it is probably working satisfactorily
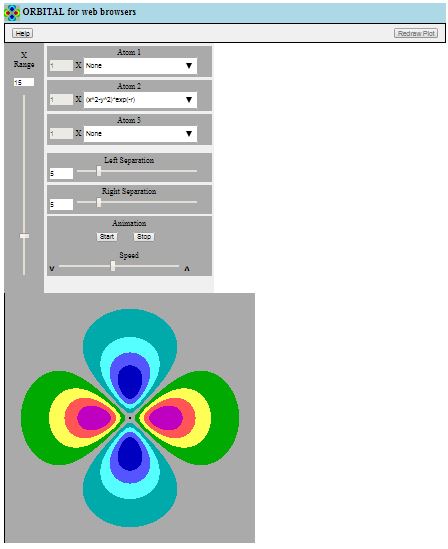
In a narrow window, it may appear folded, as shown here
This may also work, though possibly less conveniently
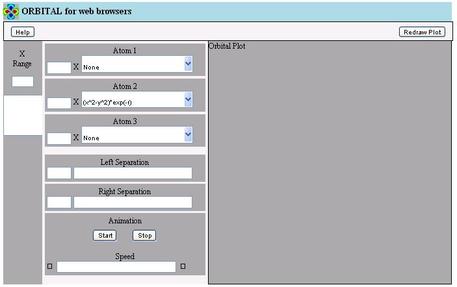
- If ORBITAL comes up with blank controls and/or without a contour
plot, as shown in the bottom screen shot, then your browser does not
support enough of HTML5.0, and cannot be used for ORBITAL
for Web Browsers
- The shot shows IE8. It is known that versions of IE before 10
cannot be used. IE11 works, but there are problems with the
accuracy of the mouse over the controls
- It may be possible to obtain a more recent browser for your device
What is it for?
-
ORBITAL is an aid to teaching and learning about molecular orbital theory
in chemistry courses at an elementary undergraduate level
What does it do?
-
ORBITAL shows Atomic Orbitals or Molecular Orbitals on a computer screen,
as coloured contour plots
-
It provides simplified mathematical functions which are of the same form
as the space-dependent parts of some of the one-electron wavefunctions
for an atom
-
Atomic functions can be shown, or functions can be added to make MOs for
a diatomic or a linear triatomic molecule
-
The program is a tool for students to use
-
It was originally devised for use in a drylab situation in which students
were taught interactively by a demonstrator while they carried out loosely
specified exercises using ORBITAL
-
It can be used in private study by students, but probably should be accompanied
by a tutorial or a quiz provided to fit in with the course they are studying,
suggesting work to do using ORBITAL
-
Teachers possibly could show it as a lecture demonstration
-
The student controls what the program does
-
There is no tutorial or programmed learning nature to ORBITAL, beyond what
is provided in this help facility
Introduction Contents
Realism
-
Although the functions have been simplified by removal of almost all of
the combinations of constants which look confusing in textbooks, the resulting
plots are very similar to those obtained with properly calculated AOs
-
This emphasizes that understanding the algebraic form of AOs is within
the grasp of almost all university chemistry students
-
Commercially available modelling packages may produce more accurate surfaces
(or possibly contour plots) for orbitals, but the algebraic form of the
functions used is generally hidden from the user
-
If used for teaching or learning, the learning objectives are different:
these packages are not meant to promote understanding about how MO theory
works, whereas ORBITAL sets out to do this
-
Ready-made rotatable models
of atomic orbitals
or
of diatomic molecular orbitals
are among the excellent web publications of Dr. P. Falstad,
but these also list the orbitals by name, rather than giving the functions
used
Introduction Contents
Recognising the AOs
-
Students select the atomic wavefunctions by their algebraic form, rather
than by their name
-
This exercises students' understanding of the algebraic nature of natural
orbitals, nodes, etc.
-
Wavefunctions are given in the Cartesian coordinate system, rather than
in polar coordinates
-
The author finds that students find it much easier to understand and assimilate
these functions, and it is anticipated that they will be taught about them
in this form
-
Molecular modelling using MO methods, as used by chemists, employs Cartesian
coordinates
-
Polar coordinates were used in the middle years of the 20th century, before
the computer era, when students learned to solve Schroedinger's Equation
for the hydrogen atom by hand
Introduction Contents
Plane of the plot
-
Atomic orbitals are plotted in the xy plane: thus 3d(xy)
and 3d(x2-y2)
orbitals are provided, but 3d(z2)
is not
-
The molecular axis must therefore be the x axis, not the z
axis which is the convention
-
The x axis is horizontal across the screen, y is vertical
down the screen (and z, which is not used, comes out of the screen
towards the user)
-
The nuclei lie in the plane of the plot, and are represented by black dots
Introduction Contents
Colour of the contours
-
The contour bands are coloured with warm colours green to purple to represent
positive values of the functions, and with cold colours cyan to deep blue
to represent negative values
Introduction Contents
Nodes
-
Values near zero are coloured neutral mid grey, so nodal surfaces intersecting
the plane of the plot are easily recognised
-
Students should exercise their knowledge of both AOs and MOs by identifying
and counting the nodes in the plots
-
The idea that the wavefunction has zero value at a node is of infinitely
little importance to chemistry, because the nodal surface is infinitely
thin
-
What is of supreme importance is that the wavefunction changes sign at
the node: see the help item on animation
-
Nodes are not shown as infinitely thin in ORBITAL, because the student
needs to see them
-
The coloured bands represent equal ranges of the value of the wavefunction,
and the grey band spans zero symmetrically (except in the 1s orbital),
making it wide enough to see easily
Introduction Contents
Making MOs
-
MOs can be made by adding one AO at a time from each atom
-
Sufficient AOs are provided to make σ bonds from s or p
orbitals, or π bonds from p or d orbitals
Introduction Contents
Coefficients
-
Students enter coefficients for each AO
-
By varying the signs of these coefficients, bonding or antibonding MOs
can be made
-
Students thus learn to use the rule that the sign of the product of two
coefficients must match the sign of the overlap integral of these two AOs,
in order to produce a bonding contribution to the MO
-
They should set out to produce a bonding or antibonding combination in
each case, and confirm by counting the nodes in the MO that they have obtained
the intended outcome
-
By varying the magnitudes of the coefficients, polarised MOs can be produced
-
Students should decide in advance which nucleus in the plot is to represent
which nucleus in the molecule
-
By examining the shape and colouring of the lobes, they should confirm
that the polarisation shown agrees with their assumptions about relative
electronegativity of the atoms, in both the bonding and the antibonding
MOs
-
Antibonding MOs are polarised in the opposite direction to a corresponding
bonding MO
Introduction Contents
One overlap at a time
-
Because understanding bonding or antibonding contributions is a main learning
objective in using the program, use of more than one AO at a time per atom,
e.g. for s and p orbital mixing in σ MOs, is not supported
Introduction Contents
Distance between the atoms
-
The distance between the atoms can be varied, showing the progression from
almost pure AOs to highly bonding or antibonding MOs, as the overlap integral
becomes bigger in magnitude
Introduction Contents
Triatomic molecules
-
The important ideas which can be explored with the linear triatomic molecule
are:
-
σ and π MOs each delocalised over all three atoms
-
the non-mixing of s and p on the central atoms, because they
require different combinations of signs of coefficients of the ligand orbitals
-
there are π MOs which are neither bonding nor antibonding: they
are non-bonding by symmetry
-
ORBITAL's triatomic molecule does not bend
-
Understanding what happens to MOs if the molecule bent is an interesting
and important topic, but it is more advanced than is usually covered in
courses at the level which ORBITAL is meant to support
-
As the symmetry is lowered, more mixing of AOs into MOs takes place, and
the situation becomes rapidly more complicated
-
The simple approach of ORBITAL, in which only one
orbital at a time per atom is considered, becomes too limiting
Introduction Contents
What are the molecules?
-
Since ORBITAL does not set out to make a realistic
plot for a particular molecule, this is a matter for the imagination of
the student or their teacher
-
Diatomic molecules could be H2, F2,
O2, NO, or HF
-
Triatomic molecules could be CO2 or N2O
-
The triatomic combination could be part of a larger molecule:
-
two trans ligands surrounding an octahedrally coordinated metal
-
the metal-carbon-oxygen sequence of carbonyl complex, in which the d-π*
π-bonding can be depicted
-
Rotatable ab initio models of MOs, for comparison with the
ORBITAL plots, are available
for CO and
for CO2
-
While a 3p(x) orbital is provided to support learning about
inner lobes, 2s is not provided
-
Because atoms do not approach sufficiently for major overlap of the inner
lobes of their AOs, it is suggested that 1s and 2p AOs are
used in constructing MOs, even where 2s, 3s, or 3p
orbitals are involved in 'reality'
-
This gives MOs which are of the correct form in the bonding region, but
which are oversimplified in the core regions near the nuclei
-
This approach is often taken in elementary textbooks to avoid unnecessary
confusion
Introduction Contents
Animation
-
Our ideas of σ and π bonding, or of bonding, non-bonding and antibonding
orbitals, which have carried over from small inorganic molecules to the
most symmetrical of organic molecules, are all about nodes, i.e. the sign
of values of the wavefunctions at different points in space
-
Students often need to work hard to lose the concept of electrons in atoms
as particles, which is very harmful to their understanding of chemistry,
and to replace it with the concept of electrons as wave phenomena
-
What is being plotted in ORBITAL, and in elementary textbooks, is the space-dependent
part of the wavefunction, i.e. a snapshot at some instant in time of a
parameter which varies in a wavelike manner
-
Lobes on opposite sides of a nodal surface have opposite signs because
the wave phenomenon is out of phase in these two regions
-
It does not matter which region contains positive and which contains negative
values, because a snapshot at a different instant could have shown the
signs reversed
-
This is illustrated by the animation provided in ORBITAL, in which the
space-dependent wavefunction set up by the student is multiplied by a sinusoidal
time-dependent function
-
This causes values of the total wavefunction, and hence the colours and
extents of the contour bands to change periodically, from the maximum values
shown in the static plot, down through zero, and up to the maximum values
but with the opposite signs
-
Students are advised to turn down the speed of the animation until they
fully understand what they are seeing
-
The animation may have some reality for single-electron atoms in a steady
state: for polyelectronic atoms, the time-dependence is very much
more complex
-
The animation is provided mostly to encourage students to think about the
meaning of the static space-dependent plots, which are similar to those
shown in most elementary textbooks on the subject, but which are frequently
misunderstood by beginners
Introduction Contents
History of ORBITAL
- ORBITAL has been used for teaching first year Chemistry students
the basics of LCAO for every Honours intake year since 1976
-
The present author wrote the ancestor of ORBITAL in 1976 in Hewlett-Packard
Basic
-
Students accessed a time-shared computer via a 10-characters per second
teletype and a telephone linkage
-
The program printed different upper-case letters to represent contour values,
with white paper in between to aid visibility
-
A plot took about 12 minutes to print
-
Students changed function definitions by editing the program before running
it
-
The idea came from L.J. Soltzberg, J. Chem. Educ., 1972, 49, 357-361
-
By 1985 the program had been translated into various dialects of Pascal
for use in stand-alone microcomputers, but was still being used in an edit-compile-run
mode
-
Monochrome screen output still used letters to represent contour values,
but was a lot faster than teletype output
-
In 1993, Dr. Chris M. Smith assisted in translating ORBITAL into Prospero
Pascal, and used its colour graphics capability to produce coloured contours
similar to the present version
-
A sequential output-input conversation was used to set up all required
parameters
-
The general nature of the previous versions, in which students could enter
any function (in Pascal), was sacrificed in favour of selection from a
menu of predefined functions in a pre-compiled program
-
In 1999, the ProPascal version became unusable because of purchase of PCs
with non-compatible graphics adapters
-
In 2003, a decision was made to translate to a stand-alone Windows
application, using ClearWin+ Fortran, because of the greater ease of using
interactive
controls and particularly of implementing user-determined animation
-
In 2018 a translation was made to use web browser-based HTML controls
and the JavaScript language
Introduction Contents
Using ORBITAL
-
ORBITAL loads with an atomic orbital already selected
-
This may be changed using the drop-down function list for atom 2, or the
X Range may be changed
-
Any change except setting the Animation on,
results first of all in the plot being greyed, to show that it no longer
corresponds to the selected settings
-
Most details of the plot can still be seen, to help judge what new settings,
e.g. Range, are required
-
Once the plot has been greyed, the controls respond quickly, so that e.g.
the type-in box for Range keeps up with movement of the slider below it
-
Small buttons may be provided by the browser in use,
at the side of the type-in boxes: these are 'spin
wheels': if they are kept pressed with the left mouse button, the
value is stepped by pre-set amounts
-
When the plot is greyed, the Redraw Plot button at the top right of the
display is ungreyed
-
Press Redraw Plot to remake the plot using the new settings
-
X Range controls the square xy region to be plotted
-
A higher value makes the orbital seem further away
-
Beware of using too low a value, i.e. zooming in too far, as this could
result in essential features of the orbital being outside of the field
of view
-
The same units are used for Range as for the interatomic Separations, but
because the supplied functions are simplified,
these are arbitrary: they do not correspond to Ångstrom units
nor to Bohr radii
-
Animation may be stopped by clicking the Stop button
-
Because animation stops at an arbitrary magnitude, the plot is left greyed
and Redraw Plot should be pressed
Atomic Orbitals
-
Atomic wavefunctions are plotted if any one of the three atoms has an algebraic
function selected and the other two have None
-
The coefficient typein boxes are greyed, and even if they are set
already, they have no effect
-
The interatomic Separations have no effect
Molecular Orbitals
-
Molecular wavefunctions are plotted if any two, or all three of the three
atoms have algebraic functions selected
-
The bond direction is horizontal, i.e. along the X axis
-
Use 2p(x) for σ bonding or antibonding functions and
2p(y) for π bonding or antibonding functions
-
Left separation is between atoms 1 and 2, and right separation between
2 and 3
-
If functions are selected only for atoms 1 and 3, the sum of the two separations
may be used to select a much larger interatomic distance: e.g. use
a separation of at least 20 units, made in this way, to display σ bonding
using 3p(x) functions
-
Separations of 3 units may be suitable for π bonds using p orbitals,
or for bonds using the 1s orbital
-
A separation of 3 units is too short for σ bonds using 2p orbitals:
separation 5 units is suitable
-
The 3p(x) function is supplied mainly for display as an atomic
orbital (use a large X range)
-
Remember to increase X Range to include the separation selected as well
as enough range for the lobes themselves
-
Selecting too large an X Range can easily be corrected, but selection of
a too small range may lead to wrong conclusions
-
Coefficients may be typed in, or, if 'spin-wheels' are provided,
these may be used to adjust the current value
-
The range of these spin wheels is less than may be typed in
to the boxes
-
For polarised bonds, if coefficients are set to above 1 for some atom(s)
and below 1 for the other(s), the range of the spin wheels is sufficient
to represent any reasonable degree of polarisation
- Clicking to the left of a coefficient allows a minus sign to be inserted,
but some browsers may not provide a minus sign for numeric input
- Because of this, a ± button is provided for each
coefficient
- This conveniently reverses the sign of the coefficient, and
should work in all browsers
Modification
- The code of ORBITAL for Web Browsers is entirely client-based,
so may be used offline, if the available browser allows that
- This means that the program is essentially open-source, since a user
can view and save all of it, using the View Source facility of the
browser
- A user with sufficient programming skills could modify the code,
e.g. to change the list of functions provided
- If ORBITAL is republished online, modified or not, it would be
reasonable to acknowledge its original author
Feedback
I should like to hear about your use of ORBITAL, and be sent suggestions,
comments, etc. I do not undertake to
act on suggestions, but I certainly welcome them and will give them due
consideration.
Thanks very much.
Bruce Tattershall
Chemistry in the School of Natural and Environmental Sciences
University of Newcastle
Newcastle upon Tyne
England
Email: Bruce.Tattershall@ncl.ac.uk
Website: http://www.staff.ncl.ac.uk/bruce.tattershall/