Select the format in which molecular structures are to be shown:
- JSmol requires HTML 5.0, and can be slow
- Jmol requires Java to be installed on the client machine, but is sometimes much faster
Format currently selected: HTML5.0_JSmol
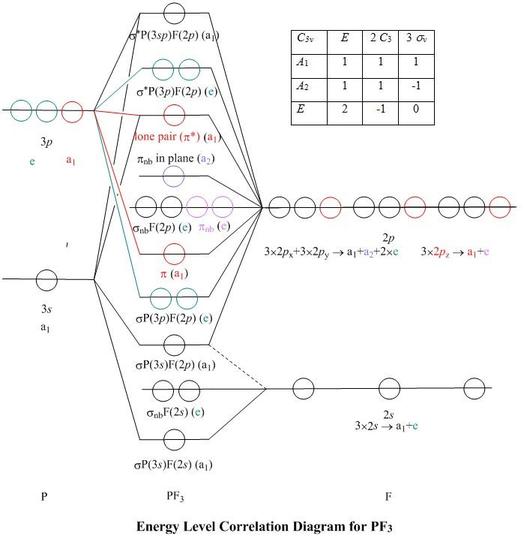
Molecular Orbitals for PF3
Jmol models of wavefunctions calculated at the RHF/3-21G* level
To view a model, click in the circle of a molecular orbital in the energy level correlation diagram shown
- Ignore any popup warning and click on the green Continue button which appears
The energy level diagram may be displayed with or without the group theory symbols and
character table: the models accessed by clicking are the same.
Group theory symbols are currently onMouse Control of Models
Left mouse drag to rotate; Shift Left drag up or down to resize;
Shift Right drag or Shift Left drag horizontally to z-rotate;
Right click for menu
Notes
Usage
- The orbitals models are shown in two popup windows, which are reused alternately so that
you can compare one orbital with another
- Contours on a two-dimensional plot correspond to surfaces in three dimensions
- The initial view of a model is with surfaces at ψ = ±0.04
- A radio button is provided to 'Switch contours on'. This shows a
two-dimensional contour plot in a plane
- This plane is xy or yz or xz, chosen so that you see some contours for each model.
This is the plane of the screen when the model first appears, though you may then turn
it around
- This calculation is
slow in the default HTML5.0_JSmol format for the structures. It goes much more
quickly if you can use a Java-enabled browser such as IE11 and can switch to the
Java_Jmol format using the button provided at the top of this page.
The calculation of the surfaces is acceptably fast in either format
- You may see the contour plot better if you 'Switch surfaces off' and click
'Contours coloured'. Black and white contours are provided as the default because
the coloured ones do not show up very well through the coloured surfaces
- If you 'Switch ball and stick off' you can see contours right up to the nuclei,
and for some MOs you may see inner lobes with opposite phase
Symmetry and sp Mixing
- Planar
BF3
has D3h symmetry, and different symmetry representations for
B 2s and 2p orbitals, so they do not mix in any of its MOs.
PF3, in contrast, because of the pyramidal conformation at phosphorus, has
only C3v symmetry, which is a subgroup of D3h
- This means that the P 3s orbital belongs to the same a1
representation as the P 3pz orbital, so they can mix in all
the MOs which have a1 symmetry
- Correlation lines to both AOs have been marked in on the Energy Level Correlation
Diagram only where both make a significant contribution to the MO
Molecular Orbitals and their Labels
- I have named the MOs on the energy level diagram and
in the titles in the model windows according to the more major AO contribution
from phosphorus and/or fluorine
- Most inorganic chemists
probably consider that in pyramidal PF3,
the π system of planar BF3
should have entirely given way
to σ bonding and antibonding orbitals,
together with the lone pair on phosphorus
- However, if one examines the MO labelled π and shown in red on the energy
level diagram, it looks remarkably like the π bonding orbital of BF3,
except with a curved π nodal surface, instead of planar, to accommodate the
pyramidal group of atoms
- Consequently, I have named the orbitals 'sigma' or 'pi' on the energy level diagram and
in the titles on the model windows, following where appropriate the classification
used for BF3
- To aid comparison,
a separate web page is provided, showing models of orbital surfaces for
corresponding pairs of PF3 and BF3 orbitals
- The MO labelled 'lone pair', which is the HOMO of PF3, is weakly
antibonding, and is the antibonding member of the set of π MOs marked in red on the
diagram
- Both the set of fluorine 2px and
2py AOs and the set of fluorine 2pz AOs
reduce to give a a1 representations,
so mixing of px,
py and pz fluorine orbitals can give resulting
p–type lobes at any angle to the z axis, in the a1 MOs
- In contrast to the π bonding MO, where the nodal planes of the fluorine p
contributions are aligned with the bond directions to produce the π node of the
MO, there is very little fluorine 2px and
2py contribution to
the lone pair MO. The fluorine p–type lobes in the MO are nearly
parallel to the z axis and the orbital might better be described as σ*
rather than as π*
- Clearly, it is a phosphorus sp mixture orbital, with a visible
p-type inner lobe, and contains, according to the present calculation,
37% P(3s), 32% P(3pz) and 9% from each fluorine
2pz
- The highest energy valence-shell MO, labelled as
σ*P(3sp)F(2p), has more phosphorus
p–character, and contains 19% P(3s) and
43% P(3pz). The rest is practically all fluorine
2px and 2py, so the fluorine
p–type lobes point perpendicular to the z axis
- The doubly degenerate pair of e symmetry antibonding MOs, shown in green
on the Energy Level Correlation Diagram, can contain no phosphorus 3s
character. They are the LUMOs of PF3
- Seen down the z axis of the pyramid, these MOs resemble the
doubly degenerate pair of π* LUMOs of
carbon monoxide, seen along its molecular axis
- Similarly, the lone pair HOMO of PF3 resembles the weakly antibonding
HOMO of CO. The similarity of these frontier orbitals results in a similarity
of PF3 to CO as a π–acceptor ligand in transition metal chemistry
MO Calculation
- These orbitals were calculated at a low ab initio level which can, however,
show bond polarisation and fully delocalised molecular orbitals
-
The method optimises orbitals filled in the ground state molecule, i.e. up to the HOMO
-
Unoccupied orbitals, i.e. the LUMOs and upwards, are devised with the correct symmetry
and to be orthogonal to the filled orbitals, but at higher energies they appear to become
progressively less like combinations of valence-shell Slater-type atomic orbitals
- The MO optimisation was followed by a Natural Bond Orbital analysis which
allowed the MOs to be described as containing the various percentages of Natural
Atomic Orbitals reported in the discussion above