Select the format in which molecular structures are to be shown:
- JSmol requires HTML 5.0, and can be slow
- Jmol requires Java to be installed on the client machine, but is sometimes much faster
Format currently selected: HTML5.0_JSmol
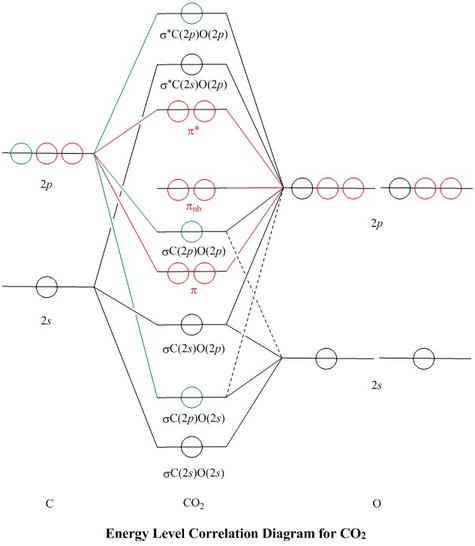
Molecular Orbitals for CO2
Jmol models of wavefunctions calculated at the RHF/3-21G* level
To view a model, click in the circle of a molecular orbital in the energy level correlation diagram shown
- Ignore any popup warning and click on the green Continue button which appears
Mouse Control of Models
Left mouse drag rotate; Shift Left drag resize; Shift Right drag z-rotate;
Right click for menu
Notes
- This web page presents evidence from an ab initio
modelling calculation, which may be useful to those
learning or teaching about molecular orbitals for
simple inorganic species
- It does not set out to teach
- If you would like a tutorial aimed at students
following a beginners' course about atomic orbitals
and their linear combination to make molecular
orbitals, you could try
Tutorial using the program Orbital
sp Mixing
-
The σ orbitals (green or black) lie symmetrically across the π nodes of the π orbitals
(red), so the two do not mix
- The node of the C(2p) orbital (green) coincides with an element of symmetry of the molecule,
so C(2p) (green) does not mix with C(2s) (black): they stay orthogonal
in the molecule and contribute to different σ MOs
- In contrast, the nodes of the O(2p) orbitals (black) do not coincide with an element of symmetry of the molecule,
so O(2p) does mix with O(2s): they are not orthogonal in the molecule.
Both s and p orbitals of O are coloured black on the diagram. Both contribute to some extent to all of
the σ MOs, though only the more important correlation lines are marked in.
For a further exploration of sp mixing, see
Molecular Orbitals for CO
- Non-mixing of s and p orbitals of the central atom is characteristic
of the symmetric molecular geometries:
symmetric linear triatomic, trigonal planar, tetrahedral and octahedral
MO Calculation
- These orbitals were calculated at a low ab initio level
(rhf/3–21g*) which can, however,
show bond polarisation and fully delocalised molecular orbitals
- At the much higher level df/6-311g(2df) the calculated molecular orbital models
look very similar, but the weakly antibonding
MO σC(2p)O(2p) appears below the bonding π MOs in the energy
level diagram