Web Pages for Teaching and Learning Chemistry
by Dr. Bruce W. Tattershall, Chemistry, Newcastle University, England
This is a list of web pages designed for undergraduate students
of Inorganic or Structural Chemistry
Note about Popups
- Most of the pages in this list show calculated models
in popup browser windows or tabs,
so that the user can compare two models
- Most browsers block popups, so when you click on a
chart or button to select a model, you will see
a warning box rather than the desired model
- There is no need to reduce the security of your browser,
because a green
Continue
button will also appear on
the web page, near to the chart or button which you clicked
- If you are working in a small window, you may need to
scroll a bit to find this green button
- Click the green Continue button.
This should satisfy the browser that you are not malware,
and the popup should open, showing the desired
model
Molecular Orbitals
Web Application Orbital
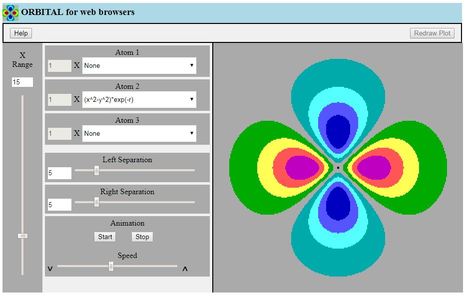
This is a two-dimensional contour plotting program intended for
students following an introductory course about atomic and
molecular orbitals
- The student selects from a list of simplified functions
expressed in Cartesian coordinates, which serve to represent
natural atomic orbitals
- The program plots coloured bands on the screen, to represent
either one atomic wavefunction, or two or three of these
wavefunctions added together to make a molecular orbital
- The student appreciates the idea of a wavefunction, and learns
about nodes in AOs and in MOs, and hence about π versus
σ MOs and antibonding versus bonding MOs
- LCAO coefficients and nuclear separations (on arbitrary scales)
can be set at will as experiments
- The program is best used in a classroom situation with a script
designed by the instructor to fit the course being given, but the
original online tutorial script used in Newcastle with the
Orbital application has now been updated and
is available online
Pre-calculated Molecular Orbitals for Simple Molecules
This series of web pages provide on-screen rotatable molecular
models, with their valence-shell molecular orbitals, taken from real
ab initio calculations. Species range from homonuclear
diatomics, e.g. peroxide ion, to PF
3, a trigonal
pyramidal molecule with π-bonded ligands
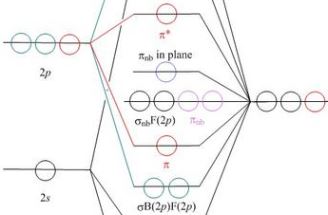
- All of these pages work in the same way, using the same or
similar web application software
- Each has a schematic molecular orbital energy level correlation
diagram, and when the student clicks on a MO, a new window pops up
to show a JSmol/Jmol model of a surface of the selected orbital
- The models come up as a ball and stick model of the geometry, with a surface at
ψ = ±0.04 for the selected orbital, coloured blue
and red to show relative signs of the wavefunction
- A radio button is provided to switch on a contour plot in the plane of the screen in the
initial view. For each orbital the direction of view is selected so that some useful
contour plot is visible if the contours are switched on
- What is different between these pages are the notes provided,
which point up the principles illustrated by the particular species
Molecular Orbitals of Peroxide Ion
[O2]2–
This a rather more familiar example of a formally singly bonded
homonuclear diatomic species, than the isoelectronic fluorine molecule,
F2
- From a Natural Bond Orbital analysis of the calculated MOs,
tables of LCAO coefficients of natural atomic orbitals in
the MOs, and of contributions of their overlaps to bonding,
are provided and discussed
- Orthogonality of the σ and π systems
with each other, and between the π orbitals, is introduced
- sp mixing in the σ system of peroxide is compared with the N2
case, where it is much more important and, in contrast to
peroxide, it results in crossover
in energy of the σN(2p)N(2p) and the bonding
π levels
- For peroxide it is pointed out that although the
σO(2s)O(2s) MO contains
only 5.6% p character,
2s — 2pz overlaps
in it still provide 30.3% of the total bonding
- The unusual situation that the HOMOs are antibonding, and
that their negative contribution to bonding exactly cancels the
positive contribution of the π-bonding orbitals,
is dealt with
Molecular Orbitals of Nitrogen, N2
For this molecule, the subject of sp mixing is explored in
more depth
- The ab initio calculations were done at the same level
as for peroxide ion or for CO (see below), so the tables of LCAO
coefficients etc. may be compared directly, between these web
pages for diatomic species
- A button is provided to switch on an additional explanation
of sp mixing in terms of stepwise LCAO to make
σg(s) and
σu(s), etc., followed by mixing of
σg(s) with
σg(p) to produce the calculated
overall coefficients. This follows the approach in some
introductory textbooks
- The competing requirements of different natural atomic orbitals,
in determining the equilibrium bond length, is discussed.
The dominant π bonding in N2 needs a shorter
bond length, so it is too short for optimum
N(2pz) -
N(2pz) overlap
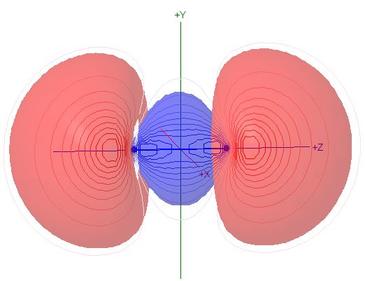
Molecular Orbitals of Carbon Monoxide, CO
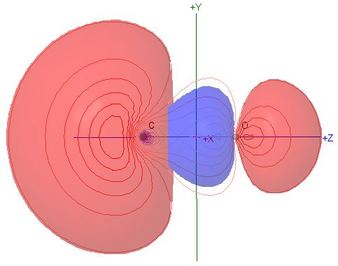
Polarisation of molecular orbitals is the chief new topic for this
heteronuclear molecule
- The percentage compositions of most MOs, in terms of the
contributions of natural atomic orbitals, are shown against the
correlation lines of the energy level diagram
- All of the valence shell NAO contributions to the
σ MOs are shown in the Table of Coefficients,
so the student can assess the effect of polarisation,
particularly in comparison with the isoelectronic N2
- Although the MO names are retained from the N2
case for the sake of comparison, the HOMO
σC(2p)O(2p), illustrated here,
has 56% s character, and is 90% composed of carbon NAOs
- The LUMO π* orbitals are seen to be
polarised towards carbon, with 71% C2p
character, and the concept of synergic bonding as a
π-acceptor ligand to a transition metal is mentioned
- A switch is provided to select either a high level of
calculation for comparison with N2, or a
lower level of calculation for comparison with CO2
(see below). Both the JSmol/Jmol models available,
and the tables and notes, change appropriately on clicking this
switch, but the student may see relatively little difference
in either
Molecular Orbitals of Carbon Dioxide, CO2
For this larger molecule, there are much less detailed notes.
The topics to be introduced for CO2 are as follows
- Both σ and π MOs are each delocalised over
all three atoms
- s and p orbitals on the central atom do not mix
with each other, because they require different combinations
of signs of coefficients of the ligand orbitals.
Correlation lines and orbital symbols on the energy level
correlation diagram are colour coded accordingly
-
There are delocalised π MOs which are neither bonding nor antibonding,
but are non-bonding by symmetry
- The LUMO π* orbitals are strongly
polarised towards carbon, corresponding to the Lewis acidity which
is an essential property of CO2
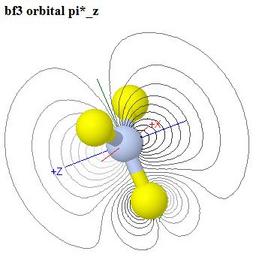
Molecular Orbitals of Boron Trifluoride, BF3
This web page is meant for a course at a slightly higher level than
is the page on CO2, so while a similar colour coding of
the energy level correlation diagram is used as in the
CO2 case, optional group theory symbols and a
character table are given as well
- These may be turned off using a provided switch, if a
suitable group theory course has not yet been taken
- As in the CO2 case, the 2s orbital on
the central atom does not mix with any of its 2p orbitals,
because they have different symmetries relative to the molecular
symmetry
- There are five delocalised MOs which are fluorine 2p
combinations that are non-bonding by symmetry
- The LUMO π* orbital is even more strongly
polarised towards boron than the antibonding LUMOs are towards carbon
in CO2, and BF3 is a strong Lewis acid
Molecular Orbitals of Phosphorus Trifluoride, PF3
How do molecular orbitals change, in going from the trigonal planar
molecule BF3 to the trigonal pyramidal molecule
PF3? A simple answer might be "surprisingly
little"
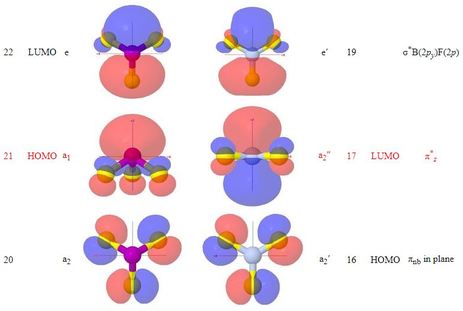
- Corresponding MOs are sufficiently similar in appearance, between the
two molecules, that the names devised for BF3 MOs have
been carried over to those of PF3
- The web page shows optional group theory labels for the MOs
in the same way that the BF3 page does
- The point group for PF3 is a subgroup of that
for BF3
- With the loss of the planar symmetry of BF3,
the phosphorus 3s orbital belongs to the same symmetry
representation as the phosphorus 3pz orbital,
so they do mix
- While the π-bonding orbital of PF3
looks similar to that of BF3, though with the π
nodal surface bent to accommodate the pyramidal group of atoms,
the π-antibonding orbital might be better labelled as
σ*, and is the HOMO 'lone pair' of
PF3
- It is noted that the doubly degenerate LUMO pair of
σ*P(3p)F(2p) orbitals
resemble, when seen down the z axis, the LUMO pair of
π* MOs of carbon monoxide, and
reference is made to the somewhat parallel chemistry of
PF3 and CO as π-acceptor ligands
Pairwise Comparison of MOs of PF3
and BF3
Rather than the student selecting corresponding MOs on the two
separate web pages and seeing them in two separate pop-up windows,
this web page allows a pair of models to be selected by means
of a single click on a line of a graphical comparison table.
The selected rotatable JSmol models then come up
side by side at the bottom of the table, on the same web page
- Thus, as in the part of the table illustrated here,
the HOMO of PF3 may be
shown alongside the LUMO of BF3
- In contrast to the separate pages for the molecules, where
Java Jmol may be selected if the student's browser is
Java-enabled, only HTML5.0 JavaScript JSmol models are provided
in the comparison page, and no contour plot option is available
for the models
- A fast, modern browser should be used (at the time
of writing this, 2019, this could be Chrome in a PC, or Safari)
Molecular Orbitals of Diborane, B2H6
Learning about LCAO MO theory is usually done in the context of small
molecules, e.g. N2, CO or CO2. People doing
wavemechanical calculations on larger molecules most often seek
molecular geometries or energies of reaction or of activation, or perhaps
other observables such as NMR shieldings. They are comparatively
unlikely to look at details of the delocalised molecular orbitals involved
in these calculations
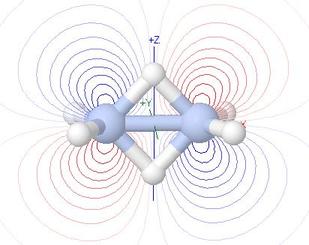
So what is the biggest molecule in which it is worthwhile to learn in
depth about the makeup of its MOs, as an aid to understanding MO theory?
- I should say it is B2H6, with 14 valence shell
atomic orbitals and hence 14 valence shell MOs
- This is because it
is a beautiful example of the application of group theory to the
understanding of orbitals: many of the nodal surfaces of the MOs
are planes of asymmetry in their group theory representation
- To make most use of this web page, a student should have a working
ability to set up a representation of a set of atomic orbitals in the
molecule, given the
character table for the point group, and to reduce this representation,
at least using a publicly-available web page to do the reduction arithmetic
- The discussion provided does use symmetry representation labelling,
on the assumption that this will be familiar to the student
Interesting Molecules
The point of providing JSmol/Jmol rotatable models
of molecular structure for
teaching and learning inorganic or organic chemistry
is usually so that the student may explore
molecular shape, conformation, chirality, or symmetry.
Physical ball and stick models, which the student may
hold and manipulate, are possibly best, but ready-made
computer graphics models have the advantage of speed and ease
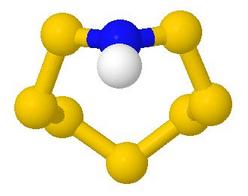
Main Groups Inorganic Ring and Cage Molecules
This web page was developed to support a course which taught about
synthesis, structure and bonding, symmetry and chirality
in non-metal ring and cage chemistry, particularly that of sulfur
and of phosphorus, where it is quite a dominant feature
- The course was a vehicle for teaching principles of bonding and
electron counting, aromaticity, and exercising the students'
ability to spot symmetry elements and hence assign point groups
- It introduced the idea of molecular chirality and its
connection with symmetry operations, beyond the introductory
organic chemistry idea of chiral centres
- Most of the examples are of academic interest only, though
some have a long history
- The web page takes the form of a table with a line for each
example
- Most lines contain one or two questions, telling the
student what to look for, and providing reinforcement of
points made in the lecture course
- Each line has a Model button, which replaces the table page
with a page showing the JSmol/Jmol model. The student
uses the back button to return to the table
- All model pages have buttons to control the appearance of
the model, and some have extra, customised controls, e.g. to
set particular viewpoints helpful to the student
in their exploration
- Each model page has an Answers or Comments button which
produces a half-width popup page, usually answering the original
questions and often giving further guidance about looking at the
model. Where the model might provide a useful example for
assigning the point group symmetry, this answer is usually given,
so as to provide a self-study tool for the student
Reaction Pathways
Traditionally, reaction pathways have been represented in
illustrations by
structural formulae of initial and final states, connected by
reaction arrows via transition states or intermediates.
Lecturers may demonstrate the well-practised manipulation of
lecture-room-sized ball and stick models, but it is difficult for
students to make adequate notes, and, when bond making and breaking
is involved, the task of delivering a good demonstration
approaches the superhuman. Smooth graphical animations, going
from one recognisable state to another, are an excellent teaching
and learning replacement for physical models. Alternative
representations, e.g. spacefilling versus ball and stick, can
be provided easily if desired, and the student may rotate the
model on their screen so as to see the animation from any
viewpoint
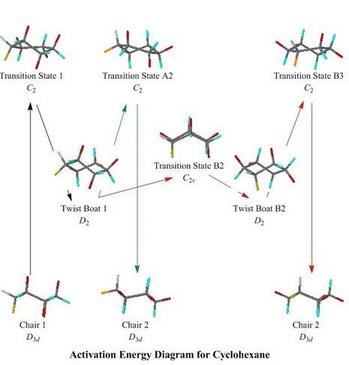
Ring-Flipping of Cyclohexane
This is probably one of the first fluxional molecules that
chemistry undergraduates meet, as well as being an historical
starting place for conformational analysis
- The default JSmol mode works adequately in a fast browser
such as Chrome in a PC or in Safari, but students with a
Java-enabled browser may select the Java_Jmol mode to obtain
faster animations and response to mouse rotations etc.
- Like the
MO web pages above, this Cyclohexane web page has a clickable
schematic energy level diagram, but this time showing the end
conformations, intermediates, and transition states in the
inversion of the cyclohexane ring
- Clicking on any of these structures produces a JSmol/Jmol
rotatable model in a popup window
- Up to two such windows are allowed, so that two different
structures may be compared
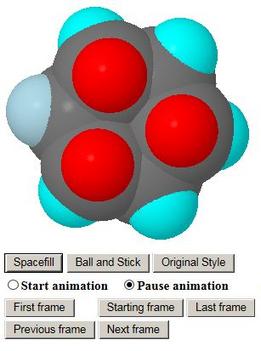
- The models come up as stick models and in profile so that
e.g. the chair shape of the end conformations may be seen
- The student may turn them around or select the more exciting
representations Ball and Stick or Spacefill
- Axial and equatorial hydrogen atoms are colour-coded so
that the student may follow them during the animation
- When all is ready, radio buttons allow the animation to be
switched on or off, and further buttons allow a quick move to
the beginning or end of the reaction pathway, or to nudge
backwards or forwards
- A frame counter allows the student to keep track of where
the animation has got to, and this shows also the name of
each of the clickable structures if and as they are passed
- Quite a lot of notes are given on the calling web page,
suggesting experiments for the student to do and explaining how
to find the symmetry elements
- Besides the simple reaction path via a Twist-Boat intermediate,
given in most introductory textbooks, this web page shows also a
'Route B', going from one Twist-Boat intermediate to its
enantiomer, via an untwisted boat transition state
- The clickable models came from ab initio optimisations,
and many of the intermediate frames from ab initio IRC
calculations. A few were interpolated by hand
Rearrangement of a Chiral Main Groups Cage Molecule
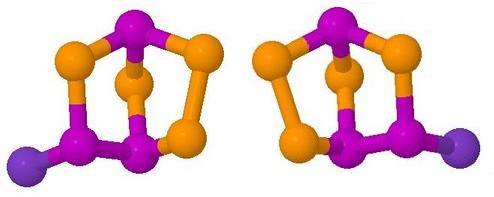 This is one of the
'Interesting Molecules' referred to above.
It is an obscure phosphorus-selenium cage molecule which
undergoes a racemisation equilibrium reaction on the
dynamic 31P NMR timescale at room temperature.
This involves breaking and making of cage bonds
This is one of the
'Interesting Molecules' referred to above.
It is an obscure phosphorus-selenium cage molecule which
undergoes a racemisation equilibrium reaction on the
dynamic 31P NMR timescale at room temperature.
This involves breaking and making of cage bonds
- To see the animation of the reaction path,
click on the Animation Model button
- This animation always starts at the transition state, since
the student needs to come to terms with that first
- Now click the Start animation radio button
- As in the cyclohexane case above, when the end of the reaction
path is reached, the animation reverses and goes backwards
- All of the points were from an ab initio IRC calculation
- For ease of the ab initio calculations, the model used
for the animation was P3Se4Br rather than
P3Se4I.
The bromide undergoes a similar exchange reaction
but at a slower rate than does the iodide
Substitution Reactions in Metal Complexes
Each of the four following web pages deals with a substitution
mechanism in transition metal complexes which goes
via an intermediate.
As in the cyclohexane web page above, there is a clickable
reaction scheme, but in this case it is the reaction arrows
which are clickable, rather than the end states of the reaction
steps
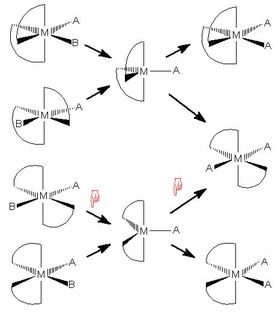
- Each page contains a reaction scheme with reaction arrows
leading to or from one or more intermediates.
Clicking on a reaction
arrow produces a JSmol/Jmol model positioned beneath the
reaction scheme
- One or two models side by side are allowed:
the left model is for a reaction leading from the starting
material to the intermediate,
while the right model is for the step going from the intermediate
to the product
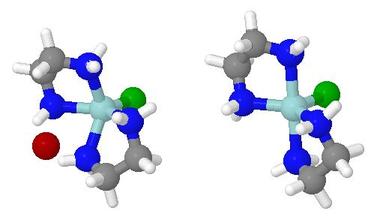
- When an arrow is clicked, a red hand symbol indicates which
step has been switched on
- Clicking the provided Animate button switches the program
to animate mode: starting with the leftmost model, a series of
animation frames are shown, representing the course of the
reaction. If two models have been selected, then when the
left model ends at the intermediate, the right model starts
automatically, carrying on from the intermediate to the product
- The models come up with their own Pause and Play and
positioning buttons, so the student can stop and reposition
the models at any point in the sequence
- Fuller introductory notes and instructions are given on the
calling page, as well as buttons for selecting
Java Jmol models instead of HTML JSmol, or for selecting a
background colour, and on the individual
pages there are Help buttons
- The models are anonymous in terms of the identity of the metal
or of the attacking or the leaving groups.
In fact, the starting and end points were semi-empirical PM3
models of imaginary zinc complexes, and the leaving and
attacking groups were bromide and chloride respectively
- Animation steps were from Gaussian geometry optimisations
going towards the stable molecules, and the reverse sequence
for dissociation steps. This is explained more fully in an
About.. page giving the history of these web pages
The four constituent web pages are as follows
-
SN1 Mechanism for Substitution in an Octahedral Complex
- This goes through a square pyramidal intermediate
-
Enantiomers in a SN1 Mechanism for a Bis-chelate Octahedral Complex
- This goes through a trigonal bipyramidal intermediate
-
Associative Substitution Mechanism for a Square Planar Diethylenetriamine Complex
-
Associative Substitution Mechanism for Square Planar Complexes with Monodentate Ligands
- This shows retention of configuration, even in the absence of a constraining
tridentate ligand
Molecular Vibrations
Some molecular modelling packages can calculate the vibrational
modes of molecules, which give rise to infrared and/or
Raman spectroscopic absorptions. To understand these, it is
very desirable to see animated models which can be rotated
so as to show the most informative view
A traditional chemical application of Group Theory,
taught in undergraduate chemistry courses, is the
calculation of the number, symmetry and nature of the
vibrational modes of symmetrical small molecules.
Possibly the most difficult part of this is visualising what the
molecular motions 'look' like, particularly for
symmetry representations which are doubly or triply
degenerate. Several pages on the Web show JSmol
models of vibrations, to support such courses
Vibrations of Some Phosphorus Chloride Species
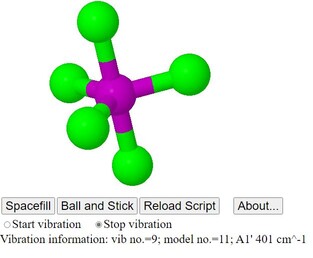
This page shows animated JSmol models of the vibrational modes
for some phosphorus chlorides which have molecular geometries
common in Inorganic Chemistry
- The models are from wavemechanical modelling, though at a
very low level
- The calculated vibration frequencies are not intended to
approximate to experimental spectroscopic values, but serve
to show the calculated order the vibrations, and which are
degenerate with which
- There is no tutorial content in this page: it just shows
the models resulting from calculations, without comment
- Users may find it useful in conjunction with learning or
teaching the application of Group Theory to molecular
vibrations